如何科学、客观地制订溶出度试验质量标准
谢沐风
上海市食品药品检验所 上海市浦东新区张衡路1500号
邮编:201203 邮箱:xiemufeng@sina.com
摘要: 本文详尽阐述了如何科学、客观地拟定质量标准中溶出度试验各参数,并廓清了拟定出发点和控制要素,为如何使该试验法的拟定体现出制剂内在品质提供了佐证与参考。
关键词: 溶出度试验法;拟定;参数
溶出度试验作为“评价固体制剂内在品质的灵魂与核心”,随着人们对该项技术的不断研究与深入,对其认识与理解亦在不断发展与变化着
[,,]。现今,该试验不仅具有为建立体内外相关性而设立的宗旨,且还已成为证明药物体内释放特性的一种简单、廉价而不失严谨的实验室检测方法。
尤其“在多种pH值溶出介质中溶出曲线(本文的“溶出度曲线”包涵释放曲线,以下同)的测定(以及比对)”更是成为“剖析”和“肢解”固体制剂内在品质一种擘肌分理、抽丝剥茧的重要手段;成为固体制剂内在品质呈现于外在的一种“表象”、“映射”与“载体”;成为在仿制药“殊途同归”的研发进程中、提高生物等效性(BE)试验成功率所历经的“必由之路”,甚**成为该品种在其工艺与处方完全成熟、持续进行多批大规模生产后,所拥有的特定特征“指纹图谱”。总之,“多条溶出曲线的测定”可在与固体制剂品质相关的所有环节上均发挥出举足轻重的作用。
无论是研发仿制药/创新药,或是省级药检所在审核质量标准、起草药典、标准提高/转正、评价性抽验、不同来源的药品间品质评价等各项工作,均涉及到在进行了大量深入的研究验证后,**终应如何科学、客观地制订质量标准中溶出度试验法,即通过该法测得的结果能否充分反映出该产品所应具有的内在优良品质、所应具有的良好体内生物利用度以及品质均一性,能否通过该试验法的制订促使企业进行深入的制剂工艺/处方研究与周而复始的品质保证等等
[,]。
截止目前,关于如何制订质量标准中溶出度试验法的文章尚未有发表。本人已撰写发表多篇了有关溶出度试验在药物研发、质控、内在质量评价等方面如何发挥作用的文章
[,,],本篇将就本人工作经验与感悟,详尽阐述如何制订溶出度试验质量标准。
一、
针对仿制药
无论是《药品注册管理办法》中的三类新药(已在G外上市销售但尚未在G内上市销售的药品)还是六类新药(已有G家药品标准的原料药或者制剂),两者**区别就是有无“可参照的质量标准”,但均请按照以下思路进行。
1.
既有质量标准的可参照性
针对六类仿制药,实验者肯定会查询到相关参考文献,如各G药典、G家药监局标准、进口质量标准、《日本橙皮书》、美GFDA公布的溶出曲线数据库、日本仿制药技术申报资料概要(采用“医薬品インタビューフォーム”在Google上收索即可,内容仅为日文)等,但由于这些标准本身所具有的局限性、历史性和利益性,因此jue不建议实验者不经对原研品的剖析(详见2~7内容),就直接撷取,即便是G外药典或进口质量标准,制订者也不应盲目迷信(详见讨论1)。这正是G家药品审评中心自2008年起就反复提出“仿产品不是仿标准”宗旨的核心要素之一(对于口服固体制剂,还有一要素为“有关物质”)。
2.
购买原研品
六类仿制药,均可购买到进口品或原研厂在G内合资企业产品。
三类新药(其实也是仿制药),研发者必须想尽办法购买来原研品,可通过私人关系带入,或通过“世界药房”网站、“白求恩药房”网站等中介,还可向G家药监局注册司申请“一次性进口”批件(少量、仅用于研发)等多种方式。jue不应以购买不来为借口,“天马行空、闭门造车”地进行处方开发与制剂工艺研究;且购买时应尽可能获取不同时间段的多个批号样品(如出厂不久的和临近效期的)。
将购买来的原研品作为0个月计,放入冰箱内冷藏。对于研发者,强烈建议与仿制品同时进行6个月稳定性考核,每一时间点样品取出后均放入冰箱冷藏,直**6个月结束。shou先测定**终时间点的多条溶出曲线(当然还有有关物质),观测与0个月相比的变化情形,再酌情考虑是否需测定其他时间点样品,从而根据原研品溶出曲线波动情况对自身仿制品的研发和品质做出正确判断与准确评估。
3.
采用多条溶出曲线“循序渐进、抽丝剥茧”般剖析原研品
在进行溶出曲线测定前,一定要先进行以下三项工作
[4,]。
3.1
“pH值-溶解度”曲线的测定
取过量原料药(可为预经微粉化处理),置8支具塞试管中,分别加pH1.0、…、8.0溶出介质适量,置37℃水浴振荡过夜,形成过饱和溶液,取出后滤过,取续滤液经HPLC法测得溶解度,绘制曲线。对于难溶性药物,对照品溶液可酌情采用纯甲醇或纯乙腈配制。
该曲线的绘制可提供众多信息:如与X轴平行,说明各pH值溶解度几近一致,由此可预测多条溶出曲线应重合;如曲线上有陡峭变化、甚**是有数量级差异,则可揭示多条溶出曲线形状必会有所差异(即高低错落),**低的那条曲线一定是溶解度**低的pH值介质,这为制剂研发方向提供了针对性与佐证素材。
当出现主成分在某pH值介质中极不稳定、溶解后即迅速分解,无法测定的情况,则该介质溶解度可不测定,其溶出曲线亦可免做。
3.2
pKa值的查询与测定
pKa值的知晓也十分重要,可通过查询或测定获得。测定法可参照以下三篇文献【,,】。若该值未涵盖于四条标准溶出曲线pH值范围,则研发时第5~6条溶出曲线就应测定该pKa值所对映的pH值介质或以上“pH值-溶解度曲线”上急剧变化的pH值,这些pH值溶出曲线的测定对于仿制制剂研发和曲线比对均具有十分重要的意义。
3.3
主成分在各溶出介质中的稳定性
为获得准确试验数据,该验证必不可少。
建议取原料药粉末配制的溶液即可,无需取样品溶出液。
3.4
采用溶出曲线“剖析”与“肢解”原研品
本人已发表此篇文章,请详见参考文献[]。
4.
质量标准中各参数的制订[4,]
在进行了以上对原研品和仿制品多pH值溶出介质中溶出曲线的测定后,才能科学客观、合理全面地制订质量标准。具体阐述如下:
4.1
溶出介质的选择
4.1.1
速释制剂
shou先应满足在该介质中**终溶出量达85%以上,然后可按下列情况分别选取
[]。
Ø 根据该药物在体内吸收主要消化道部位的生理pH值(适用于一般情况);
Ø 能在一定程度上反映体内外相关性的pH值(适用于创新药);
Ø **能体现不同来源制剂间彼此差异的pH值(适用于标准转正/药典起草、尤适合于我G大量仿制药存在情形);
Ø **能反映生产工艺变化、偏差的pH值(适用于企业内控标准,用于批间样品品质均一性的评价);
Ø 溶出曲线中**低的pH值(适用于企业内控标准,用于应对G家市场检查与监督);
Ø 溶出数据精密度更佳的pH值(某些样品在**介质中精密度不佳时、更换为精密度更为理想的介质)
当多条溶出曲线重合时(各时间点溶出量相差不超过10.0%),《日本橙皮书》倾向**“水”。其出发点为:虽然水的pH值和表面张力会因来源不同而改变,且在试验过程中也可能会因药物影响或者溶解入二氧化碳使溶出行为发生变化,但考虑到发生上述可能性的概率较小,且可通过事先验证予以探明,故秉承环保、提高试验效率等出发点,质量标准中**水。而美G药典鉴于以上问题的存在,倾向采用带有pH值的介质。笔者更倾向日本作法,因水的pH值范围5.0~7.0已被上述多条溶出曲线的pH值所涵盖。
当多条溶出曲线不重合,则可根据上述各针对性酌情拟定。
4.1.2
肠溶制剂
应规定酸中介质(pH1.0~1.2)和碱中介质(pH6.8~7.2)释放量的测定。
酸中释放量的测定现今愈发倾向通过测得准确数据予以表达、而非肉眼观察外观形状进行控制(日本橙皮书皆采用此法),通常规定2小时不得过10%。为防止药物在年轻人体内发生过量释放,甚**在该阶段可故意采用高转速(如100转),以进一步探明药物的内在优良品质。
如主成分溶出后在酸性介质中不稳定旋即降解,即便立即测定也无法准确评估时,建议测定溶出杯中剩余固体颗粒所含主成分量,随后用测得百分含量减去剩余百分含量,再除以测得百分含量,即为酸中释放百分量。
现今,随着市场上销售的肠溶衣辅料已皆可在pH1.0~3.0范围内包裹住药片,使主成分释放量合格,而经研究表明:人体内胃环境,随年龄增长胃酸分泌逐渐减少(胃内pH值范围1.2~7.6),如此再在pH1.0~1.2范围内测定已显得毫无意义,故现今开始逐步测定pH4.5介质,规定依然是不得过10%溶出量。英G药典自2008年始
[],“奥美拉唑肠溶胶囊/片”的制订原则便是以此为依据,这样的测定更有针对性和实用性,值得我们学习和借鉴!
碱中释放量同速释制剂;但需注意的是:肠溶衣对紫外测定时常有干扰,故强烈建议采用紫外-两点相减法或HPLC法,否则极易出现溶出量均值高于含量3.0%以上的情况。
4.1.3
缓控释制剂
shou先应满足在该介质中**终溶出量达80%以上。
当体外多条释放曲线重合时(酸中仅测定2小时即可),建议**水(pH5.0~7.0)作介质,既经济又方便。jue不建议采用酸性介质,因任何人体内的十二指肠**小肠消化道器官是不存在该值的;也不建议参照人体内消化器官的标准值(pH1.0~1.2、4.0~4.5、6.8)采用不同时段、不同溶出介质(不断调试)的费时费力方式、且此方式实验误差较大。
当体外多条释放曲线不重合时,建议选择**终溶出量达80%以上的、**慢的那个pH值介质。
4.1.4
介质中胃蛋白酶和胰蛋白酶的加入
一般情况下,不建议溶出介质中添加这些酶
[5]。但如某些药物必须饭后服用、且生物等效性试验需在“进食”状态下进行时,则在仿制药研发中必须进行“溶出介质中分别添加胃蛋白酶和胰蛋白酶的比对研究”,此时质量标准中亦应加入。
另:当硬胶囊剂使用囊壳为明胶胶囊时,为避免产生交联现象影响溶出,可也加入,但需进行验证。
4.2
取样时间点与限度的制订
4.2.1
普通制剂
以**次出现溶出量均在85%以上两时间点,且该两点溶出量差在5%以内时(即出现“平台期”),取前一时间点作为质量标准中取样时间点,并将该点溶出量减去15%作为溶出限度。这就将“之前我们通常认为的‘溶出度取样时间点常选择溶出曲线拐点处后推10~20分钟’
[]”的原则予以明确化和具体化,因为“拐点处后的10~20分钟”即为溶出饱和“平台期”。由此便可推断出:溶出限度一般仅有70%、75%、80%、85%四个数值。如**时间点为20分钟,由于其不为刻钟的整数倍,一般提高**15分钟,但限度可仅减去10%。
当出现采用50转、溶出曲线缓慢上升、**60分钟后才出现平台期(研究时需测定到360分钟)时,会出现取样时间点过于滞后的情形,这不利于产品的日常检验。此时为提高试验效率,可适当放宽参数(如加大转速)或采用溶出更快的介质,使60分钟前出现平台期,即不建议拟定60分钟后的取样时间点
[4]。但前提是,这些调整“不应弱化针对不同来源制剂间内在品质的区分力(如采用增加转速方式)”或“无法建立体内外相关性介质(更换的介质也必须具有体内外相关性的特质)”。
4.2.2
缓/控释制剂
应**少设定3个时间点(服用方式**2次)或4个时间点(服用方式**1次):**点为避免“突释”,应设定为试验1~2小时后或溶出量相当于标示量10~30%时间点;第二点是为考察药品溶出特性,该限度应设定在溶出量约50%时间点;**后一点是为确保药物溶出量超过80%时间点。如拟定4点,第二、三、四时间点的溶出量应分别约为40%、60%和80%溶出量。
任何一点的拟定范围均应勿超过标示含量的20%,且各点溶出限度交叉范围建议勿超过5%,除非体内特征可显示出相应的可接受性和波动性。对于零级释放产品,因其释放曲线为“一条直线”,故还应增加每小时释放量的规定(即斜率规定),“硝苯地平控释片进口质量标准(拜耳出品)”中就有6~8%/小时的严格规定。
4.2.3
治疗窗狭窄药物
为防止“突释”,愈发倾向采用两点法测定。一可采用“5或10分钟的**点溶出量不得大于某一限度(以控制突释),第二点规定一定时间内溶出不得小于某一限度以保证溶出完全”的作法(如《日本橙皮书》中卡马西平片拟定为5分钟不得过60%和30分钟不得少于70%);二可采用缓控释制剂拟定法:**点规定5或10分钟时溶出量为一限度范围而非一上限,以保证其溶出曲线呈现“缓慢上升性”(如美G药典卡马西平片规定:10分钟释放量应为30~50%,45分钟时不得少于75%),该规定亦可有效防止处方中加入大量表面活性剂或增溶剂的“投机取巧”作法。
各G制订的技术指导原则中收载的此类药物清单如下:氨茶碱、茶碱、胆茶碱、双羟丙茶碱、苯妥因钠、丙戊酸、炔雌醇/孕酮制剂、地高辛、洋地黄毒甙、华法令钠、甲磺酸异他林吸入气雾剂、卡马西平、可乐定透皮贴剂、磷酸丙吡胺、硫酸胍乙啶、硫酸奎尼丁、硫酸哌唑嗪、硫酸异丙肾上腺素、米诺地尔、扑米酮、碳酸锂、盐酸克林霉素、盐酸可乐定、盐酸普鲁卡因胺、左甲状腺素钠、环孢霉素A、他克莫司、西罗莫司、丙戊酸/丙戊酸钠等。
4.3
装置的选择
建议**通用性强、耐用性好、广泛普及的篮法与桨法。
胶囊剂**篮法、片剂**桨法
[]。
对于不易采用篮法(如发生堵塞篮法孔隙、或样品塌陷于蓝底、或粘附于篮顶)、且易漂浮于液面的制剂,可考虑采用桨法、加沉降蓝;尤对于采用了粘附性较强辅料的缓释制剂,极易发生试验过程中始终粘附于溶出杯不同部位、而使溶出数据均一性较差的情况时,更推荐采用沉降蓝。jue不建议采用自制沉降装置,因其制作的不规范性会导致数据的难以重现性。
不推荐使用非法定或非标准试验装置。如却有必要采用,但应提供详尽验证资料和充足理由,并充分验证其必要性与质量可控性(即实验室间的可操作性、耐受性和易重现性)。如美G药典第三~七法,决不能因价格昂贵而自行设计组装,且由于其现阶段该仪器尚难以推广和普及,故笔者建议勿采用。如经典的篮法与桨法无法正确表达出该样品体外溶出特征,可在此基础上进行改装,如针对栓剂或阴道制剂的体外溶出度研究与测定,即如此(详见参照文献[,])。
4.4 转速的拟定
4.4.1 既有观念的错误
由于50转的搅拌强度被看作与中老年人体胃肠道蠕动强度相当,且由于患者大多为中老人群,故**该转速。现今G内仍存在一种普遍看法,认为桨板法/50转≈转篮法/100转
[],其实这是错误的。笔者在日本G家药检所进修期间,经导师指教、并亲自采用USP溶出度校正片实验验证,结果为
[]:
l **弱级:桨板法/50转≈转篮法/50转(样品在篮内)≈转篮法/100转(样品在篮外、即沉于杯底)
l 中级:桨板法/75转≈转篮法/75转(样品在篮内)≈桨板法/50转(样品置于沉降篮内)
l **强级:桨板法/100转≈转篮法/100转(样品在篮内)≈桨板法/75转(样品置于沉降篮内)。
猜测可能是当年我G引入溶出度试验时将英文译错所致;截止目前已有不少同仁来电来函反映上式的正确性。当样品置于沉降蓝内,在试验过程中始终处于杯底,由此受到的外来涡旋力将大于样品任意漂浮或停滞于杯中某处,故其级别比不置于沉降蓝内高出一级。
4.4.2 需放宽试验参数时
不能随意采用高于50转速,因为这将极大地弱化对不同制剂/处方溶出行为的区分力。对于有必要放宽溶出度试验参数的情况,应分别考察转速为75~100转和加入表面活性剂两种方式,以更好地建立起体内外相关性和能区分不用来源制剂内在品质优劣。
目前倾向于采用“不提高转速、加入表面活性剂”方式,因对于中老年患者,虽胃肠道蠕动较弱,但体内存在胆碱类物质,可用表面活性剂表达。但当样品在杯底出现锥度堆积、呈“小山状”时,则倾向加大转速、而非加入表面活性剂方式。
表面活性剂添加浓度研究应从0.01%(w/v)起板、按照1、2、3、5级别逐步增加去“剖析”原研制剂(即逐步试验0.01%、0.02%、0.05%、0.1%、0.2%、0.5%、1.0%、2.0%和3.0%,9个浓度;另:配制溶出介质、溶解表面活性剂以及无机盐时,一定要采用加热煮沸方式,切忌采用振摇或超声),不建议采用3.0%以上浓度。当两级别间溶出曲线差异较为显著时,则应适当增加中间浓度以进一步研究。需强调的是:jue不建议不经以上“循序渐进”,而根据“漏槽条件”推断出某浓度便直接撷取(详见讨论5)。
研究时应注意不同来源的表面活性剂可能会对试验结果带来显著性差异情况,如十二烷基硫酸钠(SDS)的使用,现今愈发倾向在研究时采用市场某一主流品牌(分析级),并在质量标准中注明,以使其后试验的重现,jue不建议研究时采用市场上多个品牌、在质量标准中不注明的作法,费时费力、且导致试验结果无法预料。
综上所述,若质量标准中未制订50转或采用了添加表面活性剂的介质,就应在研发资料和起草说明中予以详尽验证和阐述。
4.4.3 **极端试验条件
质量标准中拟定的**极端条件建议为:桨板法/100转、溶出介质中加入3.0%表面活性剂。因目前上市的所有原研制剂,皆未有在此条件下没有一个溶出介质达不到85%(或80%)溶出量的产品;且在该条件下,如任何一个介质中皆未有85%以上溶出量,亦可推断该制剂犹如“石头般坚硬”,如此制剂在人体内的生物利用度也就可想而知了,这一点对于创新药的制剂研发具有很强的指导意义。
溶出介质中严禁添加有机溶剂。因为这将极大地降低体内外相关性,严重背离溶出度试验应用理念
[5],是典型的“为了溶出合格而设定”的标准;同时对于人体而言,是不可能“先喝2两白酒再吃药的”。遗憾的是,G内的一些质量标准仍有这样的品种存在,值得深思。
若确实找不到一个理想溶出条件,说明现今的制剂手段尚无法解决该原料药的溶解性,尚无法开发成适合人体吸收的制剂,此时则应果断、明智地放弃,使之休眠,直**开发出可解决其生物利用度的辅料。
4.4.4 特例
pH值特例:鉴于3.0%表面活性剂的加入,对于配制和试验操作皆会带来极大困难,此时若在pH大于8.0的介质中(即溶解度显著提高的pH值)可找到一个较为温和试验条件下**终溶出量达85%以上,则推荐采用该pH值,即便该值已背离人体正常生理值
[5]。美G药典中“阿维A酸胶囊(Acitretin Capsule)”采用“篮法/100转,pH9.6~10.0缓冲液中加3.0%SLS(十二烷基磺酸钠)作溶出介质,900ml,30分钟,85%限度”的条件就是出于该考虑。
转速特例:不推荐采用低于50转的条件,除非针对特定剂型或特定工艺。如中G药典“布洛芬缓释胶囊”拟定30rpm。此时则应格外注意仪器适用性,曾发现不同仪器间测定结果存在显著差异情况(即低转速下显示出仪器间性能差异性)。又如悬浮剂一般采用25~50rpm,但对于出现锥度堆积的情况,可通过适当增加桨速予以改善。
4.5 体积选择
统一设定为900ml或1000ml。900ml与人体消化道内的体液总体积较为接近,1000ml则是为便于计算。其他体积均无必要选择。
**于中G药典收载的第三法 —— 小杯法,是特定历史时期产物(当时液相不普及,对于小规格制剂,即便采用5cm吸收池,仍无法满足紫外吸收度应达0.2的**低要求,于是在第二法的基础之上衍生改良而来)。由于该法不满足当初设计
溶出仪时所追求的漏槽条件,更不符合大杯法所应具有的流体力学特征,故截**目前其他各G药典均未采用。现今,随着液相色谱仪在G内已完全普及,即便再小规格,亦可通过各种措施解决准确测定问题,故该法已无必要。因此,即便既有G内质量标准拟定该法,因原研制剂在研发和质量控制时从未采用过该法,故强烈建议实验者一律改为大杯法。
5.
投样量
之前曾有为满足样品**低定量限需要,采用一杯内投入2片作法,现今采用HPLC法已皆可解决
[],故投样原则必须遵循单片样品方式
[21]。
对于多计量的颗粒剂、混悬液、干混悬剂等剂型,可采用一次单位服用量投入。投入方式可采用:混匀后立即倾入已标化的、带有刻度的试管中5~10ml,即刻用滴管加样品**刻度,倾入溶出杯中,并用溶出杯中液体清洗试管。
6.
测定法[]
无论采用何法检测,均应牢固树立“研发阶段测定法与质量标准测定法应区别对待”的科学理性理念。
6.1 研发时测定法
研发时,溶出测定工作量十分巨大,具不完全统计:速释制剂仿制药研发需测定约500条溶出曲线、缓控释制剂需测定约1000条。如采用紫外法测定,就会有因需不断筛选处方而采用各种/各来源辅料对紫外测定干扰的担忧和为满足紫外检测需达0.2~0.8吸收度要求、而采取繁琐稀释步骤等的顾虑,故强烈建议采用HPLC法。关于如何高效准确、快速便捷地测定大批量溶出度样品请详阅参考文献[23],如此便可省时省力、事半功倍。
目前市售的G产“光纤全自动测定
溶出仪”、将光纤探头插入溶出杯中直接测得一条完整曲线(可每隔20秒测定),虽是采用紫外法测定,但由于其数据处理程序中已设定了各种校正法去确保排除各种情形的辅料干扰,故值得肯定与推荐。
6.2 质量标准测定法
质量标准检测仅是一个介质、一个时间点、一个限度,工作量很小,故为考虑测定的方便性,在排除辅料干扰的前提下,可考虑采用紫外法测定;但如含量测定为液相,笔者则建议采用液相,尤其是对于胶囊剂、薄膜衣片、肠溶制剂和缓控释制剂(囊壳、薄膜衣、肠溶衣、缓控释制剂辅料极易干扰紫外测定)更是如此
[]。
溶出度均值如超出含量2.0%以上,则说明有干扰,此时就会造成不合格样品误判为合格情况的出现,必须予以查明和更改测定法。目前,G内出现此种情况的品种愈发增多,盖因采用了价廉劣质的辅料所致,值得关注和警示。
7.
复方制剂
对于复方制剂,在遵循以上原则基础上灵活选择,不必拘泥于采用同一套参数。可根据各组分情况,予以针对性拟定
[]。测定法**液相。
8.
如何正确看待验证结果与既有质量标准
根据以上2~7内容 —— 如何制订溶出度试验质量标准原则,如验证结果与既有质量标准相符,则可参照采纳;如不一致,则应制订更为科学、合理的溶出度试验参数与测定法。拟定者决不能墨守成规、画地为牢,在知晓了既有质量标准不科学性、不客观性的前提下,还照搬照抄、以讹传讹!
9.
质量标准的渐进性与完善性
应强调指出的是:任何一个质量标准都不是一成不变、不可更改的!研究者可根据产品在不同阶段出现的特性和随着对该产品研究的不断深入,对质量标准中溶出度试验条件予以科学、客观的更改与完善。甚**在使用溶出度试验进行产品质量控制时,亦可根据不同情形和需要采用不同方法。
l 如申报生产时,随着制剂工艺放大和处方优化,更改申报临床时的溶出度试验参数;
l 如省级药检所拟定药典或标准转正时,可针对不同来源样品、拟定**具区分力的溶出介质;
l 如企业内控标准,为保证批间样品均一性,在完成既有质量标准规定检测的溶出介质后,增加其他更能反映批间样品波动性的溶出介质等等。
10.
溶出度试验质量标准拟定理念
由于溶出度试验是口服固体制剂的“灵魂”与“核心”所在,故如何科学、合理的拟定**关重要。其根本出发点为:制订高标准、严要求的溶出度试验质量标准来撬动制剂的深入研究,尤其是工艺药剂学的研究与实践。我G药典或G家标准中部分品种出现的“加入有机溶剂”、“150转转速”、“ 加入高浓度表面活性剂”等,皆是与以上理念背道而驰的,盖因这些品种低劣的制剂工艺所致,只好通过放宽溶出度试验条件来满足,但此种药物在人体内的生物利用度可想而知了!
二、
针对创新药[5]
创新药质量标准中拟定溶出度试验的主要目的是为避免出现那些生物利用度不佳的处方/工艺制得样品情况发生。如主成分在各pH值溶出介质中溶解度几近一致,则体外溶出行为的追求目标一定是各条溶出曲线重合。如主成分在各pH值溶出介质中溶解度相差较大,则体外溶出行为的追求目标应是尽可能地“抬高”低溶解度的那条溶出曲线,并力争使其与其他曲线重合。
shou先筛选出生产工艺和处方辅料中影响生物特性的若干个关键性因素(如4个因素),通过正交设计或均匀设计得到数个处方(如10个),小试制得各处方样品,分别进行体外溶出曲线测定,根据以上目标推断出4个关键性因素的筛选方向,按此方向,再次通过正交设计或均匀设计得到10个处方构成,再次进行溶出曲线测定,如此重复下去进行30~50个处方研究后,应可寻找到:在某一介质条件下能够区分出这些制剂溶出的优劣,即辨明“好、中、差”三个处方。然后通过动物试验(可以从小鼠过渡到大鼠,再过度到Beagle犬)验证这三种处方在动物体内生物利用度的差异性,并**好与体外溶出曲线的差异性建议起相关性(两者可不断趋同);**后规模化生产出**为优良处方的制剂进行新药临床试验验证后,按照以上思路制订该产品质量标准中的溶出度试验参数即可。
三、讨论
1.
非pH值依赖型制剂 —— **制剂的**表达。
经研究表明:随年龄增长,人体内胃酸分泌逐渐减少,正常人群胃内pH值1.2~7.6、十二指肠pH值3.1~6.7、小肠pH值5.2~6.0
[],可谓千变万化。一个优良制剂,应在人体内无论何种pH值环境下均有较为相同的释放行为,从而保证在各种环境下皆有吸收、皆有目标生物利用度。尤对于缓控释制剂,由于在体内停留时间长、“途径”各种pH值,更应具备此种“特征体外表现”。这便是制剂研发人员所应追求的“**制剂的**表达 ——体外多条溶出/释放曲线重合”目标,即应尽可能研制出“非pH值依赖型制剂”。虽然这一目标某种程度上受到溶解度或辅料性能影响,但经对众多原研品的测定表明:jue大部分药物均具有以上这些溶出特征,可见原研品在制剂研发上的投入与深入,而我G现今已上市的仿制药在该体外表观上相差甚远,其主要差距其实是对工艺药剂学的认知不足与该方面人才的匮乏。这也为我们指明了努力方向与奋斗目标!
2.
既有质量标准的局限性、历史性和利益性
2.1G内既有质量标准
由于众多历史原因和制订/审核人员当时认知的局限性,部分品种的质量标准是秉承“溶出度跟着制剂走”—— 即为了让该品种所有G内生产企业产品均合格;为了找到一个缓控释条件而体现缓控释性特性,这样一个背道而驰的错误理念制订而成。从已发表的众多溶出度研究文章和自2008年G家药监局开始实施的“全G评价性抽验所进行的探索性研究结果”中已充分体现,此处不再赘述。
2.2 英G(BP)/欧洲药典(EP)
无论是原料药还是制剂的有关物质检测,该G药典均将各杂质、色谱条件、系统适用性等关键性参数罗列得清清楚楚、一目了然;但收载的制剂却很少、口服固体制剂更少,甚**有些难溶性口服固体制剂、缓控释制剂竟然未制订溶出度检测项
[]。盖因溶出度试验作为评价口服固体制剂内在品质优良与否的核心指标,不便公开而已,故索性不予登载。
2.3 日本药典(JP)
表现形式完全同英G/欧洲药典,即便收载了溶出度试验项,也往往标注为“略”或“详见局外规”。盖因日本药品审评部门和G家药检所意识到溶出度试验重要性后,伴随1998年开始实施的“药品品质再评价工程”,将溶出度试验质量标准单独汇编成《日本橙皮书》(亦称《局外规》)单独出版。
2.4 美G药典(USP)
是各G药典中溶出度试验**具参考价值的文献,但我们亦应了解其产生过程与背景:USP一般在该药品行政保护到期前向原研企业和各仿制企业(FDA已批准、尚未上市销售)发出邀请函告知将收载该品种,希望各单位提供质量标准,当各单位提供的溶出度试验参数不同时(仿制药“殊途同归”的体现),USP不予统一、全部登载,因为这些标准均经FDA审核通过,产品皆为生物等效,这就是我们有时会看到个别品种会登载数个溶出度试验条件的原因所在。一般情况下:TSET1为原研品。如仅登载一个条件,一般也是由原研企业提供的。
然而,即便如此亦不建议制订者直接撷取,因原研品自上市到**保护期结束期间,历经大量的、各人群的临床试验验证,原研企业可能仍在不断针对处方与工艺进行优化和完善,且样品生产规模也早已远大于**初临床申报时(质量标准是那时产生),因此有些品种的质量标准已不能准确、客观地反映现今上市品所具有的内在优良品质,故笔者仍是强烈建议参照前述试验思路 —— 必须购买来现阶段上市品进行研判。如“尼莫地平片”,BP和USP皆拟定溶出介质中添加0.3%表面活性剂,中G药典完全相同,可经试验发现:原研品在0.0~0.3%溶出介质中,溶出曲线几乎没有任何变化(**终溶出量皆达85%以上,完全可以不添加),而G内众多仿制品却迥然不同:为0.0%溶出介质时,释放量很少。这充分启示我们:对原研品的质量评估不能画地为牢似地照搬照抄既有质量标准。
2.5 《日本橙皮书》[]
日本自1998年开始实施“药品品质再评价工程”以来,在《仿制药生物等效性试验指导原则》、《规格不同的口服固体制剂生物等效性试验指导原则》、《口服固体制剂处方变更后生物等效性试验指导原则》和《固体制剂改变剂型后生物等效性试验指导原则》等一系列指导原则出台的背景基础上,开始陆续出版《日本医療用医薬品品質情報集(即日本橙皮书与参比制剂目录,官方网址为:http://www.jp-orangebook.gr.jp/)》。截止2010年底,共进行了670个品种的编撰工作,其中详细罗列了每一品种的“解离常数”、“在4种pH值溶出介质中的溶解度”、“在各pH值溶出介质中的溶液稳定性”(“3. 采用多条溶出曲线循序渐进剖析原研制剂”项下已述)、“4条标准溶出曲线”、“溶出度试验质量标准”,一些品种还有“主成分对照品质量标准”、“杂质对照品质量标准”等信息。2011年1月,G家药品审评中心组织翻译了这些品种,并在其官方网站(www.cde.org.cn)主页右侧建立了《日本溶出曲线数据库专栏查询系统(中文翻译版)》
[]。
2.6美GFDA公布的“溶出曲线数据库”
美GFDA-CDER(药品审评中心)属下的仿制药办公室里的生物等效部(by the Division of Bioequivalence, Office of Generic Drugs),为提高仿制药内在品质、强化各项审评规范,从2004年初开始效仿日本,从多种溶出介质中遴选出**能反映内在品质的一个溶出介质汇编成集,登载于该部官方网站:http://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm,并于每季度更新一次。截止2011年**季度,已进行了约900个品种的披露。列举如下:
|
Drug Name |
Dosage Form |
USP Apparatus |
Speed
(RPMs) |
Medium |
Volume
(mL) |
Recommended Sampling
Times (minutes) |
Date Updated |
|
Abacavir Sulfate |
Tablet |
II(Paddle) |
75 |
0.1N HCl |
900 |
5, 10, 15, and 30 |
03/22/2006 |
同时,该部欢迎大家来电来函,对收载的参数提出异议和修改意见。关于该数据库的说明与答疑内容亦十分重要(http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm073197.htm),请读者自行浏览,此处不再赘述。
2.7 进口质量标准
由于溶出度试验的重要性,不排除会有一些企业故意放宽溶出度试验参数,从而确保进口的各批样品均符合规定的“利益性色彩存在”。这实际上也对我们的审评员和试验复核人员的技术水准和专业把握提出了更高要求。建议仍是秉承以上思路进行研判。
3.
质量标准中可不拟定溶出度试验品种和仿制药研发时可申请豁免生物等效性试验品种
口服固体制剂质量标准中并非一定要拟定溶出度检查项,当原料药属于宽治疗指数药物、并为生物药剂学分类系统(BCS)**类药物——高溶解性、高渗透性,且采用桨板法/50转,制剂可在**少四种溶出介质中均达30分钟85%以上溶出量,同时辅料量与主药量相比未超过30%,辅料中也未加入表面活性剂、甘露醇和山梨醇
[](因为这些辅料可造成溶出度试验对生物等效预测的误判)时,质量标准中就可不拟定溶出度检查,仅采用崩解时限予以控制
[]。因为此类制剂在胃内(任何pH值)的崩解、吸收已不受胃排空时间的影响。拉米夫定片(0.1g规格)
[]质量标准便如此。
当原研品属于此类制剂时,仿制药研发亦必须具有以上各特性,此时则可申请豁免生物等效性试验,不过建议溶出曲线的测定应**少为5~6条
[24]。世界卫生组织于2010年11月公布了31个此类药物清单,并对每一药物予以了详解
[]:对乙酰氨基酚(扑热息痛)、乙酰唑胺、阿昔洛韦、盐酸阿米替林、阿替洛尔、磷酸氯喹、硫酸氯喹、盐酸氯喹、西咪替丁、盐酸环丙沙星、双氯芬酸钾、双氯芬酸钠、盐酸强力霉素、二盐酸乙胺丁醇、呋塞米、布洛芬、异烟肼、拉米夫定、左氧氟沙星、盐酸甲氟喹、甲硝唑、盐酸甲氧氯普胺、强的松龙、强的松、盐酸普奈洛尔、吡嗪酰胺、硫酸奎纳定、盐酸雷尼替丁、利福平、盐酸维拉帕米、甲硝唑。
药物渗透性可通过某些网站查询,如美G口服药物传递研究公司(Therapeutic Systems Research Laboratories Inc,简称TSRL公司)网站就提供该项服务,查询网址为:http://www.tsrlinc.com/resources/services/。
4.
溶出仪的校正与校正片使用
现今,各仪器厂商皆已具备对仪器进行机械校正的能力。在这基础之上,几乎皆可通过校正片(美G/中G药典出品)测试。如无法通过,则是溶出杯问题。盖因在手工烧制底部时,无法均一性地制得内部完整半球形(烧制厚度不一所致),如图所示。此时,需更换溶出杯后再行测试。故建议使用时,安装位置、桨杆、溶出杯三者皆应统一固定
[]。
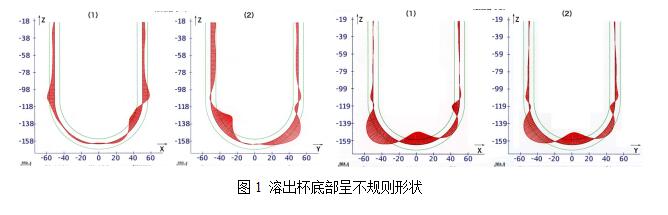
图1 溶出杯底部呈不规则形状
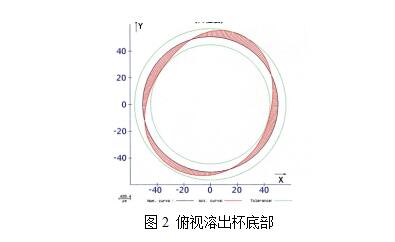
图2 俯视溶出杯底部
5.
对“漏槽条件”的再认知
漏漕条件的定义为:溶出介质体积应大于溶解药物主成分(该量为制剂**大规格量)所需体积的**少3倍量,以保证药物溶出不受其溶解性的显著影响。该理念是美G学者在设计
溶出仪设备、斟酌溶出杯体积时,根据当时已上市的大部分药物后设计的折中体积。现今,由于溶出杯体积已固定(900~1000ml),因此若以此为出发点,根据某药物在某溶出介质中的溶解度来推算采用“何种溶出介质”或“添加多少浓度的表面活性剂”,将极大地忽视“药物制剂”作用,即是对“药剂 —— 就是改善药物水难溶性程度”这一概念的摈弃。故笔者认为,该理念在现今溶出度试验应用中已无用武之地。归根结底,还是要取原研制剂,采用溶出曲线循序渐进般予以分析和辨明。
6.
需拟定溶出度检查的剂型
除通常的口服片剂与胶囊剂外,难溶性药物的颗粒剂/散剂亦需拟定溶出度检查项,因为此种剂型同样存在主成分在体内释放、吸收过程
[]。对于咀嚼片,服用时虽要求“咀嚼后吞下”,但考虑到可能有部分患者误当作普通片,故为保证该情况下药物仍能在体内快速崩散、溶出,发挥疗效,美G药典中几乎所有咀嚼片皆拟定了溶出度检查
[],而我G目前尚未制订,值得借鉴。
7.
对“质量标准中溶出度试验所发挥作用”的再认识
质量标准中的溶出度试验,由于仅是一个介质、一个时间点、一个限度的拟定。故即便经过以上严谨、周密、科学、全面的验证予以了确定,但在全面评价药物内在品质上仍会捉襟见肘、以偏概全,尤考证不同来源同一制剂品质差异性方面,更显不足。此时。则强烈推荐进行溶出曲线、乃**多条溶出曲线的测定比对工作。现今,G际卫生组织针对不同来源的同一制剂的质量监测与评价已全面引入此法
[];日本对上市仿制药的质量评价也将溶出度试验单列(其他项目仍是由地方药检所负责),由G立医药品食品卫生研究所(即G家药品检验所)药品部亲**持指导“如何采用多条溶出曲线评价口服固体制剂内在品质”工作,并与《药品品质再评价工程》的实施相辅相成
[]。
我G药监部门每年均要进行大量的市场抽查监测检验,仅按质量标准检验往往不能更深层次反映问题存在,故笔者强烈建议引入该套评价体系。自2008年,G家食品药品监督管理局开展“G家药品评价性抽验工作”以来,已将该理念与作法贯彻并实施,取得显著效果。
8.
期待与展望
制药行业作为“高科技行业”的核心体现就是制剂,具体细化**“固体制剂的工业药剂学”,而该点又是与体外溶出度试验密不可分、相辅相成的。我们只有把握住这一要点,才能“四两拨千斤”般地“一针见血”地抓住事物本质,进而推动行业发展整合与优胜劣汰。
感谢阅读此文的读者,让我们携手来夯实G产仿制药发展基石,来共同培养G产仿制药成长土壤。为了G产制剂“励精图治、杀出重围”的那**早日到来,让我们黾勉同行、砥砺同心!再次感谢您的阅读!
谢沐风. 改善溶出度评价方法,提高固体药物制剂水平. 中G医药工业杂志 2005,7(36),447~451
谢沐风. 溶出度研究系列(一).中G药品标准, 2005,6(6):42~46.
谢沐风. 溶出度研究系列(二).中G药品标准,2006,1(7):48~51.
USP33-NF28 <1092 THE DISSOLUTION PROCEDURE: DEVELOPMENT AND VALIDATION>
FIP Guidelines for Dissolution Testing of Solid Oral Products 和 FIP/AAPS Guidelines for Dissolution/In Vitro Release Testing of Novel/Special Dosage Forms*【G际药学联合会(International Pharmaceutical Federation/FIP)颁布的“口服固体制剂试验指导原则”和“新剂型体外溶出度/释放度试验指导原则”】 摘自官方网站(http://www.fip.org → Pharmaceutical Sciences and the FIP Special Interest Groups → Dissolution/Drug Release)
谢沐风. 溶出曲线相似性的评价方法 中G医药工业杂志 2009,4(40):
谢沐风、张启明等. G外药政部门采用溶出曲线评价口服固体制剂内在品质的情况简介. 中G药事,2008,3(22):257-261.
谢沐风. 提高质量标准,促进品质提升,带动行业发展 —— 议如何促进G产药品的质量 中G医药工业杂志,2007,11(38):
**亚敏. 浅谈溶出度检查方法的建立 摘自G家食品药品监督管理局药品审评中心网站【www.cde.org.cn → 电子刊物 → 20071130 浅谈溶出度检查方法的建立】
李广*,张育平等 水杨酸pKa的紫外_可见分光光度法测定. 河南科技学院学报(自然科学版)2005,1(33),75~77
樊志顺,李勤朗 用PH分配原理PKA值对临床用药问题的讨论分析 实用医技杂志 1996,2(3) 6~8
晁若冰,伍朝员,贺晓英 弱酸弱碱性药物PKA值的分光光度测定法 华西药学杂志1990,5(2) 104~106
张启明、谢沐风等. 采用多条溶出曲线评价口服固体制剂的内在质量 中G医药工业杂志,2009,12(40):946-950
青柳伸男. 日本薬局方溶出規格の設定方針について 独立行政法人-医薬品医療機器総合機構(http://www.pmda.go.jp) → 日本薬局方 → 技術情報【摘自日本G厚生省G家药品审评中心网站 → 日本药典 → 技术情报 如何制订溶出度试验质量标准/青柳伸男撰写】
用于艾滋病、疟疾和结核治疗的多来源(仿制)制剂(FPPs)预认证文件提交指导原则(补充1) 摘自世界卫生组织网站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 点“中文” → 点“继续” → 点“仿制药 (Generics)” → 点“补充1”】
英G药典2008年版
. 郑G钢 化学药品普通口服固体制剂溶出度方法验证易忽视的几个问题. G家药品审评中心-电子刊物-化药药物评价-化药质量控制 编号20071106
唐素芳. 化学药品溶出度方法研究 摘自G家食品药品监督管理局药品审评中心网站【www.cde.org.cn → 电子刊物 → 20040420/化学药品溶出度方法研究】
刘树春,刘言,范积芬. 栓剂溶出度检查方法的研究 天津药学 2003,2(15):13-16
牛丽红,陈启蒙. 卡铂栓剂溶出度实验方法研究 天津药学 2003,5(15):4-6
. 《药典注释(中G药典1990年版二部)》 1131页 化学工业出版社 1993年出版
. M. Morihara, N, Aoyagi(青柳伸男)* Hydrodynamic Flows Around Tablets in Different Pharmacopeial Dissolution Tests. Drug Development and Industrial Pharmacy, 28(6),655-662(2002)
谢沐风 高效、准确、快捷地测定大批量溶出度样品. 中G医药工业杂志,2010,7(41):
青柳伸男. 溶出試験の変動要因と適格性保証 製剤と機械, 290,2-3 (2003) 【溶出度试验注意事项与质量标准拟定的科学性与适用性】
谢沐风. 溶出度测定中应注意的若干问题. 中G医药工业杂志,2006,12(37):859-862
附件5 — 固定剂量复方制剂注册指导原则 摘自世界卫生组织网站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 点“中文” → 点“继续” → 固定剂量联合用药(FDCs)】
何仲贵等译 (美)沃特贝恩德(Waterbeemd,H.van)等著 《药物生物利用度》 化学工业出版社 2007年第1版
BP2009年版光盘 以“Prolonged-release”搜索品种名称即可。
青柳伸男. 日本のジェネリック医薬品は世界で**も厳しい基準により承認されている 月刊ジェネリック, 6,7-9 (2003)【题目:日本仿制药质量标准被认为是世界上**严格的仿制药标准】
摘自G家食品药品监督管理局药品审评中心网站【www.cde.org.cn → 新闻中心 → 工作动态→ 通知公告 → (第2页)20110128 CDE网站开通“日本药品体外溶出试验信息库”的通知】
金少鸿,宁保明译 《世界卫生组织药品标准**委员会第40次技术报告(世界卫生组织技术报告丛书)》附录7-多来源(仿制)药品:建立可互换性注册要求指导原则 中G医药科技出版社 2009年第1版。
新医薬品の規格及び試験方法の設定について 独立行政法人-医薬品医療機器総合機構(http://www.pmda.go.jp) → G際関係業務 → ICH → Quality品質/品質に関するガイドライン【摘自日本G厚生省G家药品审评中心网站 → G际协调事务(ICH) → 品质保证 《新药质量标准中各检测项目与测定方法拟定指导原则》】
G家食品药品监督管理局标准WS-802(X-588)-2002 拉米夫定片(葛兰素史克制药-苏州有限公司出品)
Biowaiver monographs 【G际药学联合会(International Pharmaceutical Federation/FIP)颁布的“可豁免生物等效性试验药物清单”】 摘自官方网站(http://www.fip.org → Pharmaceutical Sciences and the FIP Special Interest Groups → BCS and Biowaiver → Biowaiver monographs)
宁保明,张启明主译 汉森(Hanson)等著 《溶出度试验技术(第3版)》第24和45页 中G医药科技出版社,2007年第1版
谢沐风. 关于拟定水难溶性药物颗粒剂(口服干混悬剂)溶出度检查的建议 中G医药工业杂志,2006,6(37):
关于咀嚼片溶出度技术要求的探讨 药品技术评价文集(第二辑) 中G医药科技出版社 2008年出版 第64项
Theo G. Dekker等著, 作为WHO生产资格预认证(PQ)项目一部分的抗逆转录病毒产品的持续性监测 摘自世界卫生组织网站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 点“中文” → 点“继续” → 药品的监测(Ongoing monitoring)文档】
【G外考察】丁丽霞党委副书记等三人随G家食品药品监督管理局稽查局团组赴日交流(2009-10-30) “G家计划中对仿制药质量评价的溶出试验由G立医药品食品卫生研究所负责” 摘自中G食品药品检定研究院网站(http://www.nicpbp.org.cn) → 工作动态 → G际交流




